Computational Methods for Studying Microbiome Structure and Function
Computational Methods for Studying Microbiome Structure and Function
by Zach Bendiks
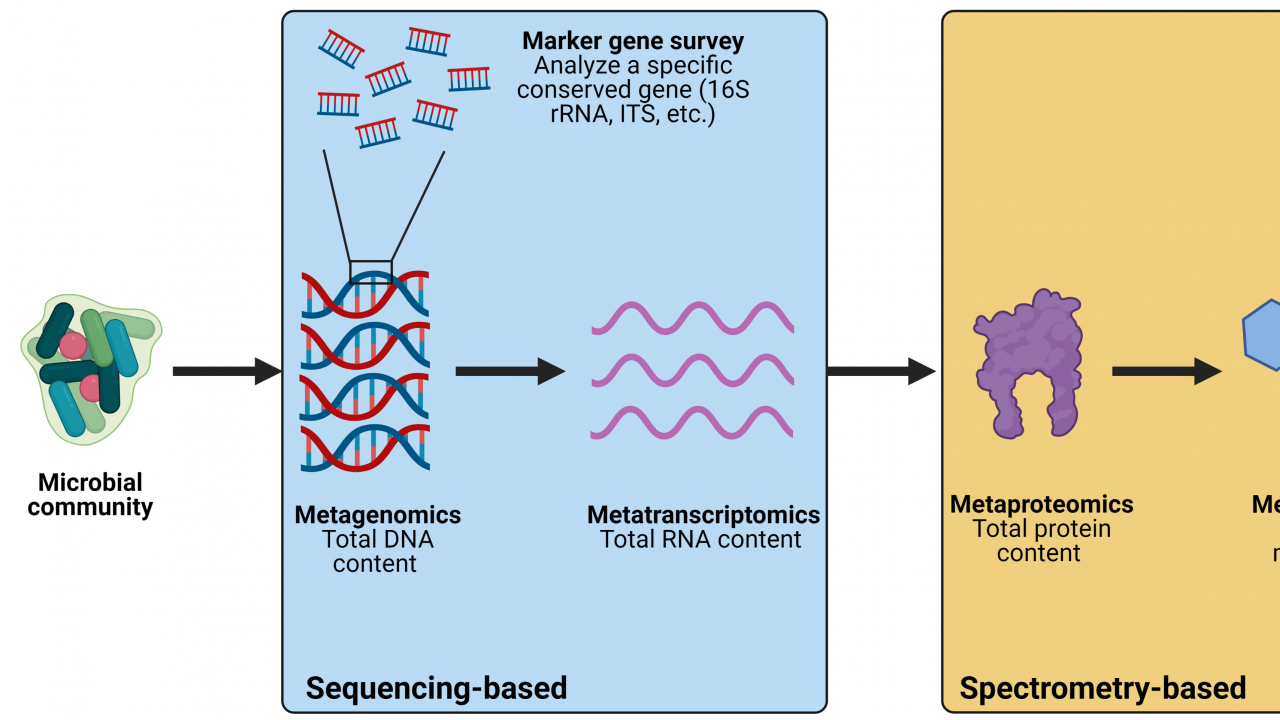
Microbial communities exist in almost every natural and human-associated environment on Earth, and these microbes play important roles in nutrient cycling, food production, and health. Because of this, much research has focused on understanding how microbial communities function in different environments and how they respond to environmental conditions. Communities present in the gut, soil, and in foods are frequently examined in research, and other communities such as those in the human-built environment (offices, bathrooms, etc.) are increasingly being studied as well. However, microbial communities often contain hundreds of different species, and many of these species are difficult or even impossible to isolate and grow in a laboratory. For decades, this was a significant obstacle for researchers who wanted to understand which microbes were present and how they functioned within a community. However, thanks to the development of modern computational techniques, we no longer rely on culturing individual bacteria but can instead gain comprehensive insight into microbial communities directly from environment samples using culture-independent techniques. In this article, we will discuss some of the commonly used techniques for studying the structure and function of microbial communities. An overview of these methods is provided in Figure 1.
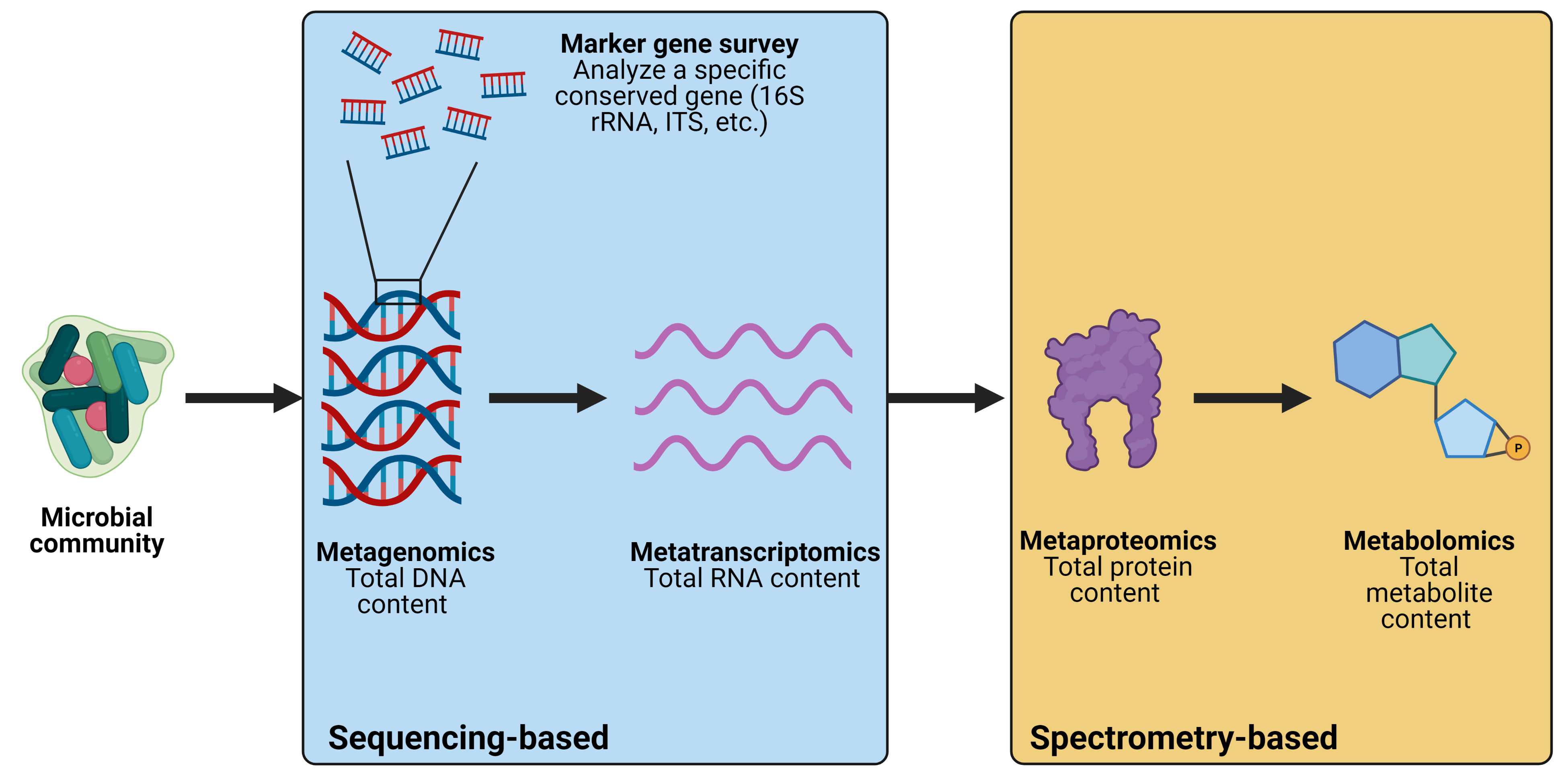
Figure 1. Overview of computational techniques for studying microbial communities. Created with BioRender.com.
DNA Sequencing-Based Methods
The development of high-throughput sequencing allows researchers to produce millions of DNA sequences that represent all the microbes present in a sample. The total DNA content of a sample can be extracted, sequenced, and analyzed computationally to figure out what bacteria are present and what genes they are contributing to the community. This is done by aligning each DNA sequence from your sample to a reference database containing thousands of sequenced microbial genomes. Analyzing the total DNA content of a microbial community is referred to as metagenomics because you are looking at the total gene content of that community. In addition, one can analyze specific marker genes within a community to identify which microbes are present. Commonly studied genes for this purpose are the 16S rRNA gene for prokaryotes (bacteria and archaea) and the internal transcribed spacer (ITS) region for fungi. All prokaryotes have a 16S rRNA gene and all fungi have ITS sequences, but the exact sequence of these marker genes vary based on the identity of the microbe encoding them. This variation allows us to identify which microbes are present within the community. These studies are referred to as marker gene surveys because the gene being examined acts as a marker that is used to identify different bacteria. Importantly, because you are only looking at a single gene and not the total gene content, you obtain less information with marker gene surveys than you would from metagenomics.
Beyond studying the DNA content of a community, one can also investigate the RNA content of a community. Metagenomics provides information about which microbes and genes are present in a community but cannot give you information about how those genes are being transcribed. This is important because an environmental stimulus may not affect the overall structure of a microbial community but may impact the function of the community by altering gene transcription. Analyzing the total RNA content of a community is referred to as metatranscriptomics because you are looking at the relative amount of RNA transcripts within a community. However, RNA cannot be sequenced directly so it must be converted into complementary DNA (cDNA) before it can be loaded into sequencing instruments. A simplified overview of the workflow for sequencing-based techniques to study microbial communities is provided in Figure 2.
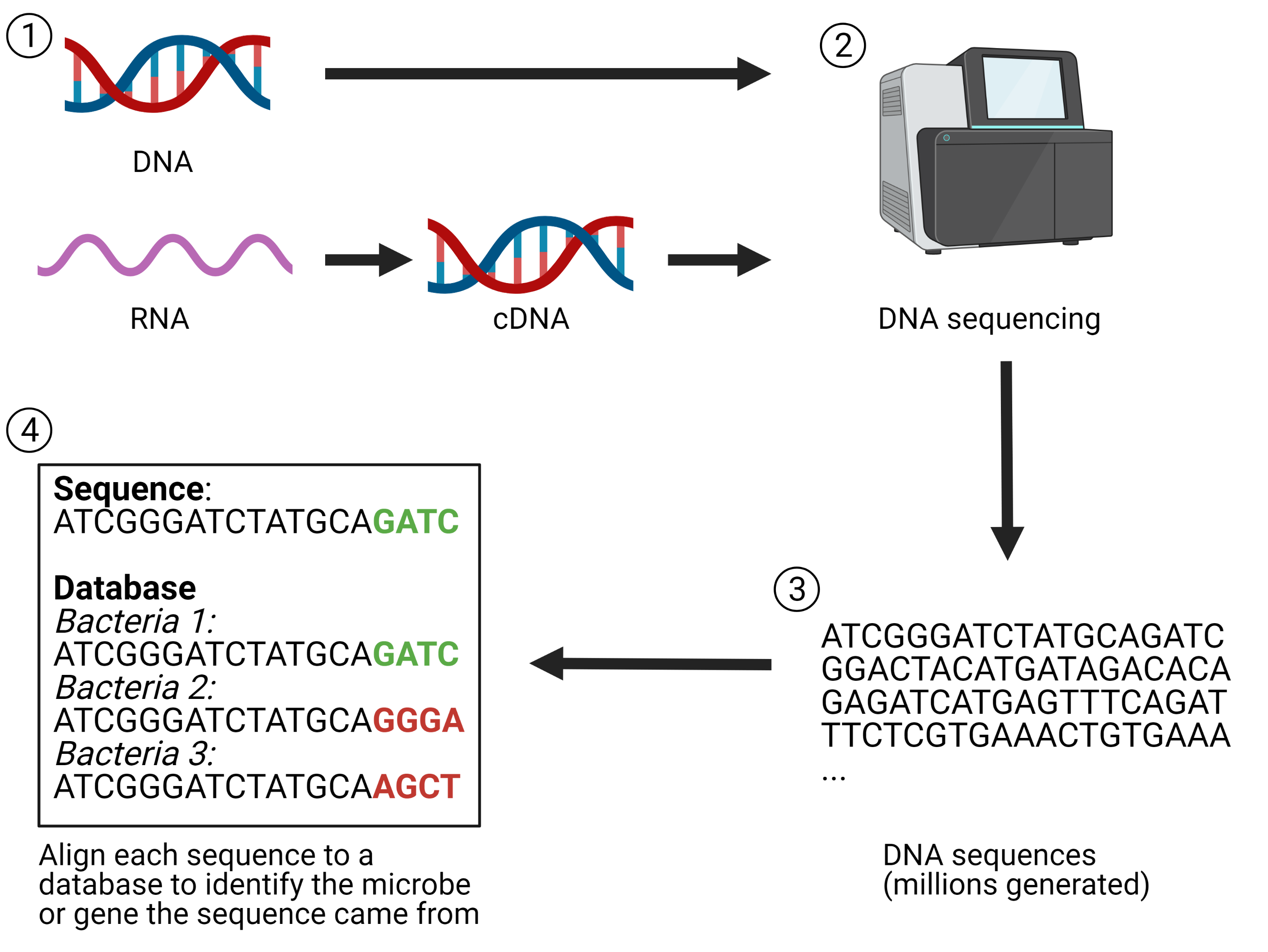
Figure 2. Simplified workflow of sequencing-based techniques for studying microbial communities. 1) Nucleic acids are extracted from microbial communities. RNA must be converted to cDNA. 2) High-throughput sequencing of the DNA. 3) Sequencing generates millions of reads, representing all the microbes present in the community. 4) Sequencing reads are aligned to reference databases containing sequences of previously characterized microbes. This allows you to identify which microbe or gene each read came from. Created with BioRender.com.
Spectrometry-Based Methods
While metatranscriptomics is a valuable tool, increased transcription of a gene does not necessarily mean the gene is more highly expressed (i.e., that there is more of the gene product). To investigate gene expression directly, one must quantify the amount of protein being produced. Researchers have developed a technique called metaproteomics which gives insight into the protein content of a microbial community. Using mass spectrometry (MS), researchers can fragment proteins that are produced by a microbial community and obtain spectral patterns for each protein. From there, the spectral patterns can be compared to a reference spectral database to identify which proteins they came from. This information can be used to quantify the expression of different genes and provide deeper insight into how the community functions overall.
In addition to proteins, microbial metabolism produces thousands of small molecules that interact with other microbes and with the surrounding environment. These molecules are referred to as metabolites and studying the metabolite profile of a microbial community is known as metabolomics. While the previously mentioned techniques look specifically at genes or gene products, metabolomics looks at the physiology of the community. Like metaproteomics, metabolomics is performed by using mass spectrometry and spectral databases to identify which metabolites are present and how their levels respond to environmental stimuli. Though some metabolomics studies have used nuclear magnetic resonance (NMR) instead of MS, the latter has several advantages over NMR and the metabolomics field has generally preferred to use MS-based methods. A simplified overview of the workflow for spectrometry-based techniques to study microbial communities is provided in Figure 3.
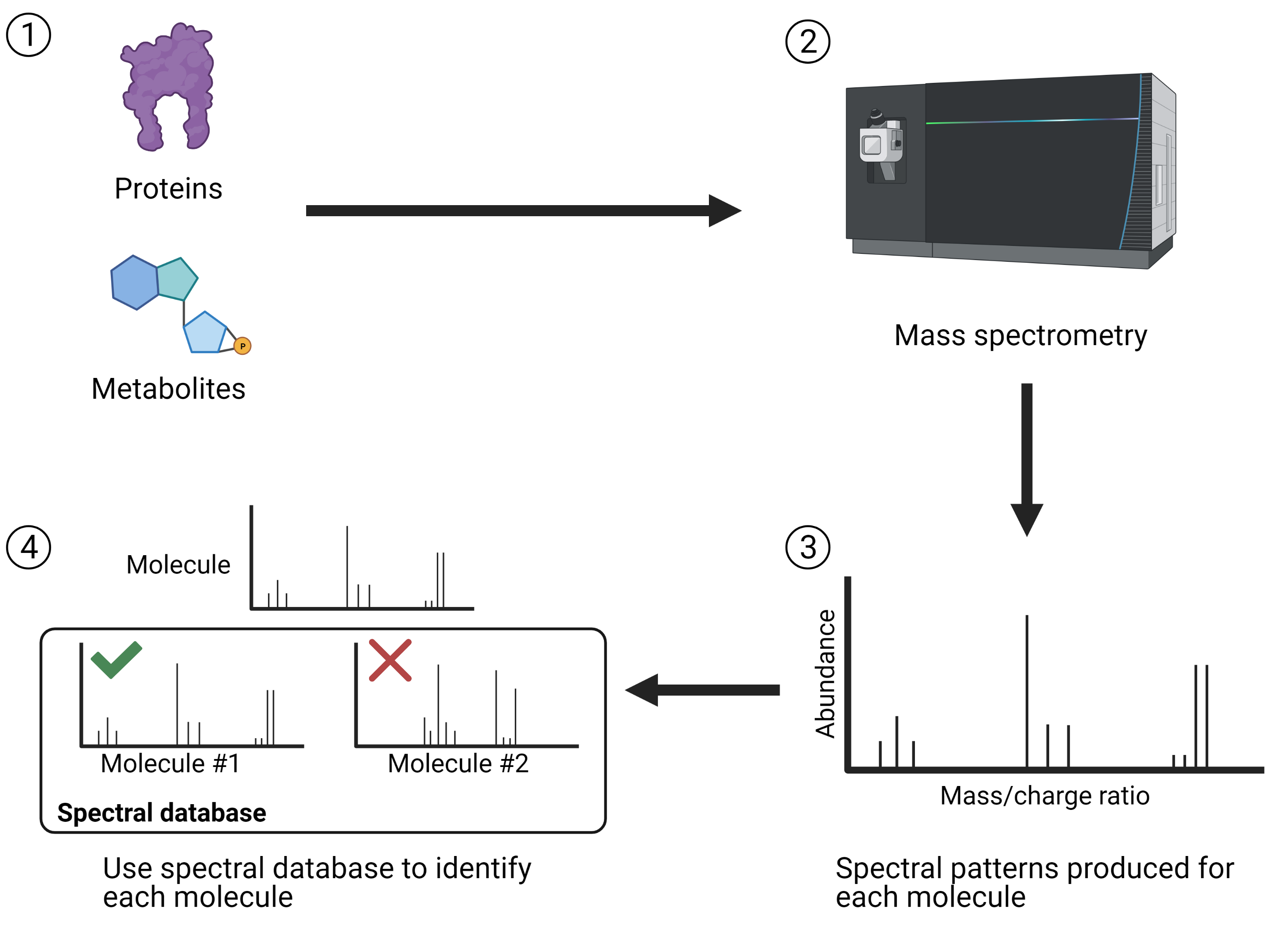
Figure 3. Simplified workflow of spectrometry-based techniques for studying microbial communities. 1) Molecules are extracted from microbial communities. These can either be large molecules (proteins) or small molecules (metabolites). 2) Mass spectrometry is used to fragment the molecules, quantify the fragments, and characterize their mass and charge. 3) For each protein or metabolite, spectral patterns are produced based on how their fragments behaved in the mass spectrometer. 4) Spectral patterns are associated with spectral databases containing known molecules and proteins. This allows you to identify which molecules are present in your microbial community. Created with BioRender.com.
Conclusions
These methods each give unique yet complementary insight into the structure and function of microbial communities, and combining these techniques can provide the greatest level of insight. For example, one study used 16S rRNA gene sequencing, metagenomics, and metabolomics to investigate how lead exposure affects the gut microbiome in rats. 16S rRNA marker gene analysis showed that lead exposure altered gut microbial community structure and reduced bacterial diversity. Metagenomics refined these results by showing that the abundance of genes associated with numerous metabolic pathways, including nitrogen metabolism and oxidative stress, were altered following lead exposure. Metabolomics confirmed the results from metagenomics by showing that the levels of metabolites associated with these pathways, such as urea and phosphoric acid, were also affected by lead exposure. Studies like this highlight how the integration of different computational techniques can give researchers more comprehensive insight into how a microbial community responds to environmental stimuli. Much effort has gone into integrating results from these different techniques, and as costs continue to decrease and new methods for integrating different techniques are developed, researchers will gain better system-level understanding of how microbial communities function.
References
Gao, B., Chi, L., Mahbub, R., Bian, X., Tu, P., Ru, H., & Lu, K. (2017). Multi-Omics Reveals that Lead Exposure Disturbs Gut Microbiome Development, Key Metabolites, and Metabolic Pathways. Chemical Research in Toxicology. https://doi.org/10.1021/acs.chemrestox.6b00401
Illumina. (2021). Identify and compare microbes from complex microbiomes or environments. Retrieved from https://www.illumina.com/areas-of-interest/microbiology/microbial-sequencing-methods/16s-rrna-sequencing.html
Peters, D. L., Wang, W., Zhang, X., Ning, Z., Mayne, J., & Figeys, D. (2019). Metaproteomic and Metabolomic Approaches for Characterizing the Gut Microbiome. Proteomics. https://doi.org/10.1002/pmic.201800363
Reuter, J. A., Spacek, D. V., & Snyder, M. P. (2015). High-Throughput Sequencing Technologies. Molecular Cell. https://doi.org/10.1016/j.molcel.2015.05.004